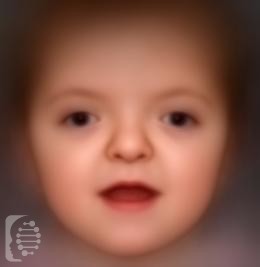
What is Apert syndrome?
Apert syndrome is a genetic condition that triggers the premature fusing of the skull bones in a child. It occurs in anywhere between 60-80,000 live births.
The main symptoms of the syndrome relate to the skull and the premature fusing of the skull bones. However, this rare disease triggers a wider variety of symptoms, affecting multiple areas of the body.
This syndrome is also known as:
Acrocephalosyndactyly – type I Acrocephalosyndactyly, Type I; ACS1 ACS I ACSI, Apert-Crouzon syndrome
What gene change causes Apert syndrome?
FGFR2 gene variants are responsible for this syndrome. The syndrome may be inherited or occur as the result of a de novo mutation. Parents with the syndrome have a 50% of passing the syndrome onto their children. Allelic disorders which share features such as Crouzon and Pfeiffer syndrome should always be considered differential diagnoses.
What are the main symptoms of Apert syndrome?
The majority of the most severe symptoms are the result of the premature fusing of the skull bones.
These symptoms include a tall skull, prominent forehead, smaller than normal lower jaw, prominent eyes, small nose and fused fingers and toes.
Other health conditions related to the syndrome include delayed mental development, vision problems, a cleft palate, recurrent ear infections, consequent hearing loss, breathing issues, hyperactive sweat glands, and severe acne in puberty.
Possible clinical traits/features:
Coronal craniosynostosis, Corneal erosion, Cutaneous finger syndactyly, Cryptorchidism, Dental malocclusion, Delayed eruption of teeth, Delayed cranial suture closure, Esophageal atresia, Facial asymmetry, Malar flattening, Ventriculomegaly, Downslanted palpebral fissures, Ectopic anus, Conductive hearing impairment, Agenesis of corpus callosum, Cleft palate, Malformation of the heart and great vessels, Cloverleaf skull, Broad distal phalanx of the thumb, Broad distal hallux, Brachyturricephaly, Bifid uvula, Convex nasal ridge, Anomalous tracheal cartilage, Acrobrachycephaly, Acne, Absent septum pellucidum, Abnormality of the fontanelles or cranial sutures, Chronic otitis media, Choanal stenosis, Choanal atresia, Cervical C5/C6 vertebrae fusion, Arnold-Chiari type I malformation, Arnold-Chiari malformation, Arachnoid cyst, Aplasia/Hypoplasia of the thumb, Aplasia/Hypoplasia of the corpus callosum, Hydronephrosis, High forehead, Broad forehead, Hydrocephalus, Humeroradial synostosis, Hearing impairment.
How is it diagnosed?
To find out if someone has a diagnosis of Apert syndrome, it is important to have a consultation and evaluation with a clinical genetic specialist. Specialists may also suggest specific genetic testing or other types of tests to help reach a diagnosis. FDNA’s AI technology can help speed up the diagnostic process by analyzing facial features and other health information.
